Genetics behind the disease

The following information serves to give background on the genetic causes and potential disease mechanisms of STXBP1 Encephalopathy as well as a brief description of the functions of the protein that is encoded by STXBP1, MUNC18-1. For more elaborate background on the molecular mechanisms of this protein please follow this link or go to the Research tab.
N: De volgende informatie dient om achtergrond te geven bij de genetische oorzaken en mogelijke ziektemechanismen van STXBP1-Encefalopathie en geeft een korte beschrijving van de functies van MUNC18-1, het eiwit dat gecodeerd wordt door het STXBP1-gen. Voor meer uitgebreide achtergrond over de moleculaire mechanismen van dit eiwit, volg deze link of ga naar Research.
STXBP1 Encephalopathy is caused by mutations in the gene STXBP1
N: STXBP1-Encefalopathie wordt veroorzaakt door mutaties in het STXBP1-gen
 Genes contain the genetic code to produce proteins. The STXBP1 gene encodes the STXBP1 protein, more commonly known as MUNC18-1. Proteins like MUNC18-1 are the molecules to make our cells and tissues work. Mutations in genes often lead to the production of abnormal proteins that cannot perform their normal function. In some cases, the effects of mutations are really subtle and the mutant protein functions almost as well as the normal protein, but in other cases the protein is not even produced at all.
Genes contain the genetic code to produce proteins. The STXBP1 gene encodes the STXBP1 protein, more commonly known as MUNC18-1. Proteins like MUNC18-1 are the molecules to make our cells and tissues work. Mutations in genes often lead to the production of abnormal proteins that cannot perform their normal function. In some cases, the effects of mutations are really subtle and the mutant protein functions almost as well as the normal protein, but in other cases the protein is not even produced at all.
In addition to the genetic code to produce a protein, genes also contain much more code, for instance, information on when to produce protein during development and/or in adulthood. This ‘non-protein-coding’ code instructs our cells to produce, for example, the MUNC18-1 protein in all nerve cells of the brain, already during early (prenatal) development until old age.
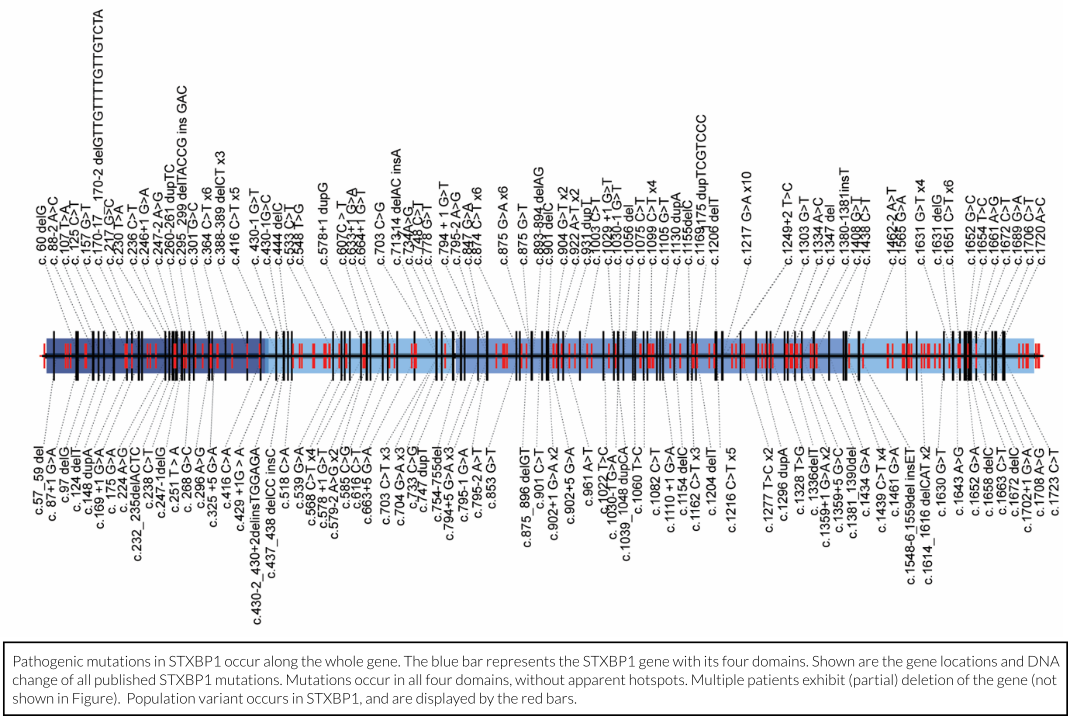
Hence, only a fraction of a gene’s genetic code encodes a protein. Most of the code is there to guide protein production and for much of the code we actually do not yet understand the function. In the case of STXBP1, mutations that cause STXBP1 Encephalopathy are usually located inside the part of the gene that encodes the protein and are found across the whole length of the gene (see figure above). In addition, mutations in the STXBP1 genetic code that does not encode the protein have also been linked to disease, especially autism, but these links are still poorly understood. Finally, several other links between STXBP1/MUNC18-1 function and brain disease have been reported, for instance, with schizophrenia and Alzheimer’s disease. Understanding such links to other diseases is one of the aims of our research.
N: Genen bevatten de genetische code die nodig is om eiwitten te produceren. Het STXBP1-gen codeert voor het STXBP1-eiwit, beter bekend als MUNC18-1. Eiwitten zoals MUNC18-1 zijn de moleculen die ervoor zorgen dat onze cellen en weefsels normaal functioneren. Mutaties in genen leiden vaak tot de productie van abnormale eiwitten die hun normale functie niet kunnen uitvoeren. In sommige gevallen zijn de effecten van mutaties zeer subtiel en functioneert het gemuteerde eiwit nagenoeg even goed als het normale eiwit, maar in andere gevallen kan het zijn dat het eiwit zelfs helemaal niet geproduceerd wordt.
Naast de genetische code om een eiwit te produceren, bevatten genen nog veel meer informatie, bijvoorbeeld over waar en wanneer tijdens de ontwikkeling en/of op volwassen leeftijd eiwitten moeten worden geproduceerd. Deze “niet-coderende” regio’s in een gen instrueert onze cellen om bijvoorbeeld het MUNC18-1 eiwit te produceren in alle zenuwcellen van de hersenen, al tijdens de vroege (prenatale) ontwikkeling tot en met latere leeftijd.
Slechts een fractie van de genetische code van een gen codeert voor een eiwit. Het grootste deel van de code is er om de eiwitproductie te sturen en van een groot deel van de code is de functie nog onbekend. In het geval van STXBP1 bevinden mutaties die STXBP1-E veroorzaken zich meestal in het deel van het gen dat codeert voor het eiwit. Deze mutaties worden gevonden over de hele lengte van het gen (zie bovenstaande figuur). Daarnaast zijn mutaties in de niet-coderende regio’s van het STXBP1-gen ook in verband gebracht met ziekte, met name autisme. Deze verbanden zijn echter nog onvoldoende bekend. Ten slotte zijn er verschillende verbanden tussen STXBP1/MUNC18-1-functie en hersenaandoeningen gevonden, zoals schizofrenie en de ziekte van Alzheimer. Inzicht in dergelijke verbanden met andere ziekten is een van de doelen van ons onderzoek.
Haploinsufficiency: A likely disease mechanism
N: Haploinsufficiëntie: Een vermoedelijk ziektemechanisme
Mutations that are predicted to have mild effects on the protein’s function (mild impact) occur just as well as mutations where no protein is predicted to be made at all (severe impact). Based on the clinical information currently available, mutations with predicted mild or severe impact can lead to the same disease severity and there is no evidence that certain symptoms are associated with one or the other. However, a more systematic comparison between predicted impact and patient symptoms is required. It is our ambition to contribute to such systematic comparisons.
N: Mutaties waarvan voorspeld wordt dat ze een milde impact hebben op de functie van het eiwit komen net zo vaak voor als mutaties waarvan voorspeld wordt dat er helemaal geen eiwit wordt gemaakt (ernstige impact). Op basis van de momenteel beschikbare klinische informatie kunnen mutaties met een milde of ernstige impact tot dezelfde ernst van de ziekte leiden en zijn er geen aanwijzingen dat bepaalde symptomen geassocieerd zijn met een bepaald type mutatie. Een meer systematische vergelijking tussen voorspelde impact en symptomen van patiënten is echter noodzakelijk. Het is onze ambitie om aan dergelijke systematische vergelijkingen bij te dragen.
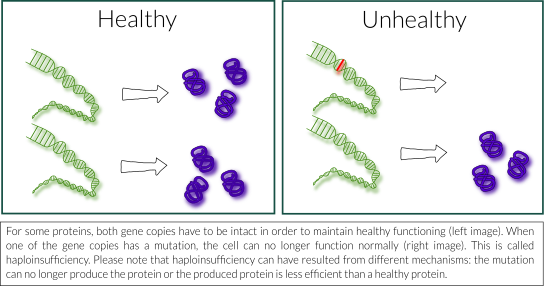
In cases where mild and severe impact mutations lead to the same disease, it is often the case that a protein that was produced from the mutated copy of the gene is not stable and is rapidly degraded inside our cells. This means that there is less protein available for the cell, as only half of the usual amount of protein is kept – the part that was produced by the healthy copy of the gene. The situation where one healthy copy of a gene is not enough to maintain healthy functioning is called ‘haploinsufficiency’. In the case of STXBP1 Encephalopathy, this would mean that half of the usual amount of MUNC18-1 protein is not enough for its normal function in our nerve cells and that this may be responsible for the disease. Haploinsufficiency was already proposed as a likely scenario in the first paper on STXBP1 Encephalopathy and is still the most likely explanation for the disease, as we also show in our paper Kovačević et al. 2018.
One of the main goals of our research is to understand how genetic mutations in STXBP1 lead to the clinical symptoms. Please visit our research page for more information.
N: In gevallen waarin mutaties met milde en ernstige impact tot hetzelfde ziektebeeld leiden, komt het vaak voor dat de eiwitten die uit de gemuteerde kopie van het gen worden geproduceerd niet stabiel zijn en snel in onze cellen worden afgebroken. Dit betekent dat er minder eiwit beschikbaar is voor de cel, aangezien slechts de helft van de gebruikelijke hoeveelheid eiwit bewaard blijft, namelijk het deel dat werd geproduceerd door de gezonde kopie van het gen. De situatie waarin één gezonde kopie van een gen niet voldoende is om gezond te blijven functioneren, wordt ‘haploinsufficiëntie’ genoemd. In het geval van STXBP1-E zou dit betekenen dat de helft van de gebruikelijke hoeveelheid MUNC18-1 eiwit niet voldoende is voor zijn normale functie in onze zenuwcellen en dat dit verantwoordelijk kan zijn voor de ziekte. Haploinsufficiëntie werd al voorgesteld als een waarschijnlijk scenario in het eerste wetenschappelijke artikel over STXBP1-E en is nog steeds de meest waarschijnlijke verklaring voor de ziekte, zoals we ook laten zien in onze publicatie Kovačević et al. 2018.
Een van de belangrijkste doelen van ons onderzoek is om te begrijpen hoe genetische mutaties in STXBP1 leiden tot de klinische symptomen. Bezoek onze onderzoekspagina voor meer informatie.
The biological function of the MUNC18-1 protein
N: De biologische functie van het MUNC18-1-eiwit

Thousands of different proteins, encoded by thousands of genes, are present in every nerve cell (neuron) of our brain. Proteins are essential for the structure, maintenance and functionality of neurons. For a neuron, one of the main tasks is to receive and send information to other neurons and to integrate information coming from different sources (other neurons, for example, in sensory organs). This communication and integration is a central aspect in the way we think, feel and move.
N: In elke zenuwcel (neuron) in onze hersenen zijn duizenden verschillende eiwitten aanwezig, gecodeerd door duizenden genen. Eiwitten zijn essentieel voor de structuur, het onderhoud en de functionaliteit van neuronen. Een van de belangrijkste taken van een neuron is het ontvangen en verzenden van informatie van en naar andere neuronen en het integreren van informatie afkomstig van verschillende bronnen (andere neuronen, bijvoorbeeld in zintuigen). Deze communicatie en integratie is een centraal aspect in de manier waarop wij denken, waarnemen en bewegen.
STXBP1 has an essential function in neuronal communication
N: STXBP1 heeft een essentiële rol in neuronale communicatie
Neuronal communication happens at specialized contact points between neurons, called synapses. It has been estimated that we have around 100 billion synapses in our brain. At each synapse, the ‘sending’ neuron conveys a message to the ‘receiving’ neuron. This works via the release of vesicles that are filled with signalling molecules, generally known as neurotransmitters. Once arriving at the receiving neuron, the neurotransmitters evoke a reaction in this cell, meaning that the message has been conveyed successfully. Within this process, many proteins work together at each step of the way. The MUNC18-1 protein, for example, plays an essential role in the fusion of the neurotransmitter-filled vesicles. Indeed, without this protein, neuronal communication cannot take place at all and no other protein can make up for this loss. In addition, MUNC18-1 has other functions in neurons and possibly in other cell types in our body. Understanding the biological functions of this protein has been a long term goal of our research team. For more elaborate information on MUNC18-1’s functions please follow this link or go to ‘Information for researchers’.
N: Neuronale communicatie vindt plaats op gespecialiseerde contactpunten tussen neuronen: synapsen. Er wordt geschat dat we ongeveer 100 miljard synapsen hebben in onze hersenen. Bij elke synaps brengt het “zendende” neuron een boodschap over aan het “ontvangende” neuron. Dit heet synaptische transmissie en dit proces gebeurt via de fusie van blaasjes gevuld met signaalmoleculen, die neurotransmitters worden genoemd. De fusie van deze blaasjes met het celmembraan zorgt voor het vrijkomen van de neurotransmitters zodat deze het ontvangende neuron kunnen bereiken. Zodra de neurotransmitters aankomen bij het ontvangende neuron, wordt er een reactie opgeroepen in deze cel. Bij elke stap in dit proces werken veel eiwitten samen om ervoor te zorgen dat de boodschap succesvol wordt overgebracht. Het MUNC18-1 eiwit bijvoorbeeld speelt een essentiële rol bij de fusie van de met neurotransmitters gevulde blaasjes. Zonder dit eiwit kan er helemaal geen neuronale communicatie plaatsvinden en geen enkel ander eiwit kan de functie van MUNC18-1 overnemen. Bovendien heeft het eiwit ook andere functies in neuronen en mogelijk in andere celtypes in ons lichaam. Het begrijpen van de biologische functies van dit eiwit is een langetermijndoel van ons onderzoeksteam. Voor meer uitgebreide informatie over de functies van MUNC18-1 kunt u terecht bij deze webpagina of ga naar ‘Informatie voor onderzoekers’.
Our research at VU Amsterdam
N: Ons onderzoek aan de Vrije Universiteit Amsterdam
Our research at the Functional Genomics Department at the Vrije Universiteit (VU) Amsterdam and Amsterdam medical center (Amsterdam UMC) has focused for many years on understanding the function of STXBP1/MUNC18-1 in nerve cells.
We have used experimental mouse and cell models in which STXBP1/MUNC18-1 protein cannot be produced due to genetic modifications that alter the STXBP1 gene in such a way that the gene cannot produce the protein (‘knock out’ technology; see figure below). It is important to note that these models do not exactly mimic the situation in (human) patients. Patients carry one healthy STXBP1 gene 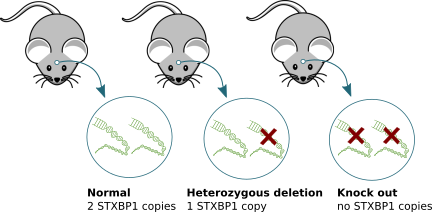 copy and one copy with a mutation, while in knock out mice and neurons both copies are defective. However, studies using these mouse and cellular models provide invaluable information on the function of STXBP1 in the brain. We have unraveled how STXBP1/MUNC18-1 organizes the initial docking of transmitter vesicles at the synapse, how it helps to prepare vesicles for fusion with the plasma membrane and with which other proteins it collaborates to do this. We have also discovered several pathways that modulate these functions of STXBP1/MUNC18-1, promoting or inhibiting its role in synaptic transmission. Please visit our research page to find out more detailed information about what our lab discovered on the function of STXBP1/MUNC18-1.
copy and one copy with a mutation, while in knock out mice and neurons both copies are defective. However, studies using these mouse and cellular models provide invaluable information on the function of STXBP1 in the brain. We have unraveled how STXBP1/MUNC18-1 organizes the initial docking of transmitter vesicles at the synapse, how it helps to prepare vesicles for fusion with the plasma membrane and with which other proteins it collaborates to do this. We have also discovered several pathways that modulate these functions of STXBP1/MUNC18-1, promoting or inhibiting its role in synaptic transmission. Please visit our research page to find out more detailed information about what our lab discovered on the function of STXBP1/MUNC18-1.
N: Ons onderzoek in de afdeling Functionele Genoomanalyse aan de Vrije Universiteit (VU) Amsterdam en Amsterdam Universitair Medisch Centrum (Amsterdam UMC) is al voor vele jaren gericht op het begrijpen van de functie van STXBP1/MUNC18-1 in zenuwcellen. We hebben hierbij gebruik gemaakt van experimentele muis- en celmodellen waarin het STXBP1/MUNC18-1 eiwit niet kan worden geproduceerd door genetische modificaties in het STXBP1-gen (‘knock-out’ technologie; zie figuur). Het is belangrijk om te realiseren dat deze modellen de situatie bij (menselijke) patiënten niet precies nabootsen. Patiënten dragen één gezond STXBP1-genkopie en één kopie met een mutatie, terwijl in knock-out muizen en neuronen beide kopieën defect zijn. Studies met deze muis- en celmodellen leveren echter essentiële informatie op over de functie van STXBP1 in de hersenen. Wij hebben ontrafeld hoe STXBP1/MUNC18-1 de verankering van neurotransmitterblaasjes aan de synaps (docking) organiseert, hoe het helpt om de blaasjes voor te bereiden op de fusie met het plasmamembraan en met welke andere eiwitten het daartoe samenwerkt. We hebben ook verschillende pathways ontdekt die deze functies van STXBP1/MUNC18-1 reguleren en daarmee zijn rol in de synaptische transmissie bevordert of remt. Bezoek onze pagina voor onderzoekers voor meer gedetailleerde informatie over de ontdekkingen die zijn gedaan in ons lab over de functie van STXBP1/MUNC18-1.
Translational approaches to STXBP1-E
N: Translationele aanpak van STXBP1-Encefalopathie
We have recently published a scientific paper where we present a mouse model that recapitulates the clinical symptoms seen in patients carrying a STXBP1 mutation. 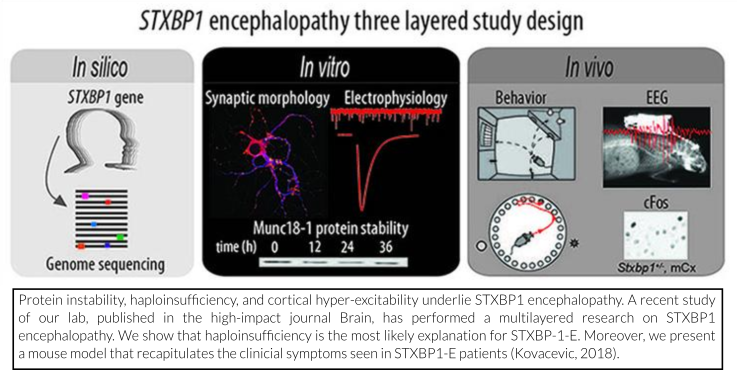
In this paper, we first provide evidence that haploinsufficiency is the most likely explanation for STXBP1-E. We show that disease-causing mutations in STXBP1 lead to reduced stability of the STXBP1 protein in nerve cells. Next, we have modeled haploinsufficiency in mice by heterozygous deletion of STXBP1. This means that one of the gene copies of STXBP1 is disabled, leaving only one healthy copy. Seizures, tonic spasms (especially during sleep) and EEG-abnormalities observed in patients with STXBP1-E are all observed in these mice. The anti-epileptic drug levetiracetam, often subscribed to STXBP1-E patients, suppressed these symptoms. In addition, these mice displayed cognitive impairments and hyperactivity. Overall, our study shows that heterozygous deletion of STXBP1 in mice provides a valid model for the development of therapeutic interventions for STXBP1-E.
For current research lines on STXBP1-E see the bottom of this page.
N: In 2018 hebben wij een wetenschappelijk artikel gepubliceerd waarin we een muismodel presenteren dat de klinische symptomen recapituleert die worden gezien in patiënten die een STXBP1-mutatie dragen.
In dit artikel leveren we ten eerste het bewijs dat haploinsufficiëntie de meest waarschijnlijke verklaring is voor STXBP1-E. We laten zien dat ziekte-veroorzakende mutaties in STXBP1 leiden tot verminderde stabiliteit van het STXBP1-eiwit in zenuwcellen. Vervolgens hebben we haploinsufficiëntie in muizen gemodelleerd door heterozygote deletie van STXBP1. Dit betekent dat één van de genkopieën van STXBP1 wordt uitgeschakeld, zodat er slechts één gezond exemplaar overblijft. In deze muizen worden epileptische aanvallen, tonische spasmen (met name tijdens slaap) en EEG-afwijkingen die ook bij patiënten met STXBP1-E worden gezien, waargenomen. Deze symptomen konden onderdrukt worden met het anti-epilepticum levetiracetam, dat vaak wordt voorgeschreven aan STXBP1-E-patiënten. Daarnaast vertoonden deze muizen cognitieve stoornissen en hyperactiviteit. Onze studie heeft aangetoond dat heterozygote deletie van STXBP1 in muizen een geldig model vormt voor de ontwikkeling van therapeutische interventies voor STXBP1-E.
Voor actueel onderzoek naar STXBP1-E, zie onderaan deze pagina.
Last update: March 6th 2023
N: Laatste update: 6 Maart 2023