A team of FGA/CNCR researchers together with US/Swedish partners (SUN project) and VU spin-off Sylics characterize the mechanisms underlying the seizures and cognitive deficits in STXBP1 Encephalopathy and present several new animal models.
In 2008, Saitsu et al. first identified mutations in the STXBP1 gene in the patients suffering from early infantile epileptic encephalopathy (Saitsu et al., 2008). In the study now published in the journal Brain, Jovana Kovačević and colleagues investigated the underlying disease mechanisms and developed animal models using a multilevel approach with in silico, in vitro and in vivo studies. The authors conclude that protein instability, haploinsufficiency, and cortical hyperexcitability together explain STXBP1 Encephalopathy. 
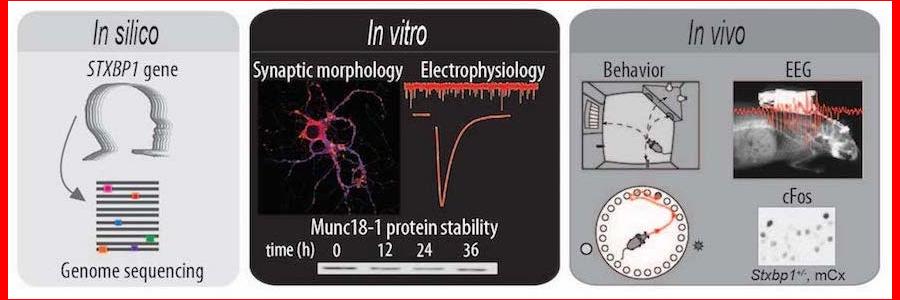
In this new study, in silico analyses of publically available genomics data of hundreds of thousands of humans show that pathogenic mutations in STXBP1 gene are ultra-rare, probably due to extremely strong consequences of STXBP1 mutations for normal brain development and -function.
The authors tested an allelic series of STXBP1 mutations in a cell culture system (in vitro) and found that all mutations lead to a decreased cellular level of the STXBP1 protein, implicating protein instability as the underlying factor to explain the disease. A previous study from the same lab has shown that lower protein levels cannot support normal synaptic transmission, at least not during intense neuronal activity. This situation is known as haploinsufficiency; normal expression of both copies of a gene is required for normal function.
 Stxbp1 heterozygous (Stxbp1+/-) mice have one functional copy of the gene and therefore provide a valid animal model for STXBP1 haploinsufficiency. The authors characterized 3 lines of Stxbp1+/- mice, on different genetic background or by deleting expression in GABA-ergic neurons only, and report spontaneous epilepsy-like behavior and abnormal EEG activity, remarkably similar to human patients. Interestingly, excessive neural activity was detected not in the brain regions most typically involved in seizure activity, but in the more superficial neocortical regions of the brain. This suggests that increased cortical excitability is the underlying cause of disease at least in Stxbp1+/- mice. New EEG analysis in STXBP1 Encephalopathy patients will elucidate whether this neocortical hyper-excitability is indeed an underlying cause in patients. In Stxbp1+/- mice the abnormal EEG activity was suppressed by the antiepileptic drug levetiracetam, again similar to the situation in humans. In addition, Stxbp1+/- mice showed cognitive deficits, especially impaired behavioral flexibility: an impaired ability to suppress previously learned responses and to learn new. Taken together, these mouse models are valuable tools for future therapy design because they model the situation in patients closely (construct validity), they show strikingly similar deficits (face validity) and respond in the same way to the same intervention (predictive validity).
Stxbp1 heterozygous (Stxbp1+/-) mice have one functional copy of the gene and therefore provide a valid animal model for STXBP1 haploinsufficiency. The authors characterized 3 lines of Stxbp1+/- mice, on different genetic background or by deleting expression in GABA-ergic neurons only, and report spontaneous epilepsy-like behavior and abnormal EEG activity, remarkably similar to human patients. Interestingly, excessive neural activity was detected not in the brain regions most typically involved in seizure activity, but in the more superficial neocortical regions of the brain. This suggests that increased cortical excitability is the underlying cause of disease at least in Stxbp1+/- mice. New EEG analysis in STXBP1 Encephalopathy patients will elucidate whether this neocortical hyper-excitability is indeed an underlying cause in patients. In Stxbp1+/- mice the abnormal EEG activity was suppressed by the antiepileptic drug levetiracetam, again similar to the situation in humans. In addition, Stxbp1+/- mice showed cognitive deficits, especially impaired behavioral flexibility: an impaired ability to suppress previously learned responses and to learn new. Taken together, these mouse models are valuable tools for future therapy design because they model the situation in patients closely (construct validity), they show strikingly similar deficits (face validity) and respond in the same way to the same intervention (predictive validity).
The original research article is available here.